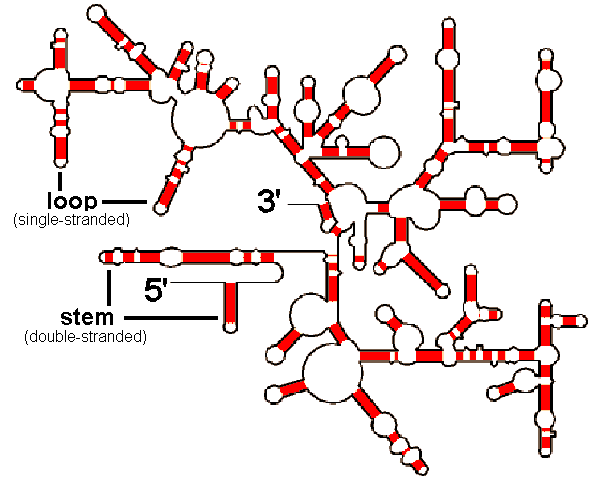
 um "copy & paste" relâmpago só para alertar que a busca pelo código-de-barras da vida (ou por uma "assinatura molecular" onde, através da análise de um trecho-específico de uma sequência de DNA, seríamos capazes de identificar espécies) tem cada vez mais suporte na bioinformática e inova ao incluir a análise da estrutura secundária de genes de RNA ribossomal (16S e outros) como caráter válido para identificação taxonômica.
um "copy & paste" relâmpago só para alertar que a busca pelo código-de-barras da vida (ou por uma "assinatura molecular" onde, através da análise de um trecho-específico de uma sequência de DNA, seríamos capazes de identificar espécies) tem cada vez mais suporte na bioinformática e inova ao incluir a análise da estrutura secundária de genes de RNA ribossomal (16S e outros) como caráter válido para identificação taxonômica. "Tradicionalmente", a análise de taxonomia molecular conhecida como "barcode DNA" ou "DNA barcoding" utiliza-se de alinhamentos da sequência primária de um trecho do gene que codifica a subunidade I da Citocromo Oxidase (COI ou cox1), especificamente, um trecho da extremidade 5' (leia-se: cinco linha) do gene. Este gene localiza-se no genoma mitocondrial (DNAmt).
obs 1: estrutura secundária: estrutura bidimensional formada a partir de interações (ligações entre nucleotídeos) da sequência primária de DNA com ela mesma (ligações "intra-fita" que geram a formação de "alças" e "grampos") .
obs 2: genes e regiões gênicas possuem orientação de leitura: de 5' --> 3'.
obs 3: a idéia de caracterizar espécies através de sequências de DNA (primárias ou secundárias) já tem longa data nos estudos envolvendo microrganismos. Mas ganhou uma nova "roupagem" e nome (código-de-barras de DNA) ao utilizar o gene COI do DNAmt para identificação de grupos animais, principalmente. Atualmente há uma tendência em minimizar a polêmica em torno desta estratégia experimental (marketing do marcador universal absoluto para identificação de espécies X defensores da falácia do "DNA barcoding" como ferramenta de identificação taxonômica) , de modo que o "barcoding DNA" tem sido reconhecido como uma ferramenta que agrega informação taxonômica válida e que tem como vantagem a caracterização de "etiquetas taxonômicas" em uma estrutura de larga-escala, baseada em uma plataforma operacional envolvendo isolamento de DNA, sequenciamento e bioinformática (bancos de dados e análises comparativas). Aliado a isso, há o esforço genuíno de integrar conhecimento taxonômico tradicional e inovações (como a digitalização e construção de amplos bancos de dados com imagens de alta resolução), incentivar a multidisciplinaridade (reconhecendo o mérito e a contribuição de taxonomistas, sistematas, geneticistas e biólogos moleculares, entre outros) e integrar a comunidade científica interessada na caracterização de biodiversidade, absorvendo críticas (o "DNA barcode" não substitui a taxonomia), abandonando paradigmas ultrapassados (taxonomia é para poucos) e buscando inovações (o artigo abaixo é um pequeno exemplo).
Veja resumo do trabalho recentemente publicado - versão eletrônica - na revista "Bioinformatics"de 13 de Abril de 2006:
Ribosomal RNA as molecular barcodes: a simple correlation analysis without sequence alignment.
Chu KH, Li CP, Qi J.
Department of Biology, The Chinese University of Hong Kong, Hong Kong, China.
MOTIVATION: We explored the feasibility of using unaligned rRNA gene sequences as DNA barcodes, based on correlation analysis of composition vectors derived from nucleotide strings. We tested this method with seven rRNA (including 12S, 16S, 18S, 26S and 28S) datasets from a wide variety of organisms (from archaea to tetrapods) at taxonomic levels ranging from class to species. RESULT: Our results indicate that grouping of taxa based on composition vector analysis is always in good agreement with the phylogenetic trees generated by traditional approaches, although in some cases the relationships among the higher systemic groups may differ. The effectiveness of our analysis might be related to the length and divergence among sequences in a dataset. Nevertheless, the correct grouping of sequences and accurate assignment of unknown taxa make our analysis a reliable and convenient approach in analyzing unaligned sequence datasets of various rRNAs for barcoding purposes. AVAILABILITY: The newly designed software (CVTree 1.0) is publicly available at the Composition Vector Tree (CVTree) web server http://cvtree.cbi.pku.edu.cn.
Chu KH, Li CP, Qi J.
Department of Biology, The Chinese University of Hong Kong, Hong Kong, China.
MOTIVATION: We explored the feasibility of using unaligned rRNA gene sequences as DNA barcodes, based on correlation analysis of composition vectors derived from nucleotide strings. We tested this method with seven rRNA (including 12S, 16S, 18S, 26S and 28S) datasets from a wide variety of organisms (from archaea to tetrapods) at taxonomic levels ranging from class to species. RESULT: Our results indicate that grouping of taxa based on composition vector analysis is always in good agreement with the phylogenetic trees generated by traditional approaches, although in some cases the relationships among the higher systemic groups may differ. The effectiveness of our analysis might be related to the length and divergence among sequences in a dataset. Nevertheless, the correct grouping of sequences and accurate assignment of unknown taxa make our analysis a reliable and convenient approach in analyzing unaligned sequence datasets of various rRNAs for barcoding purposes. AVAILABILITY: The newly designed software (CVTree 1.0) is publicly available at the Composition Vector Tree (CVTree) web server http://cvtree.cbi.pku.edu.cn.
Nenhum comentário:
Postar um comentário